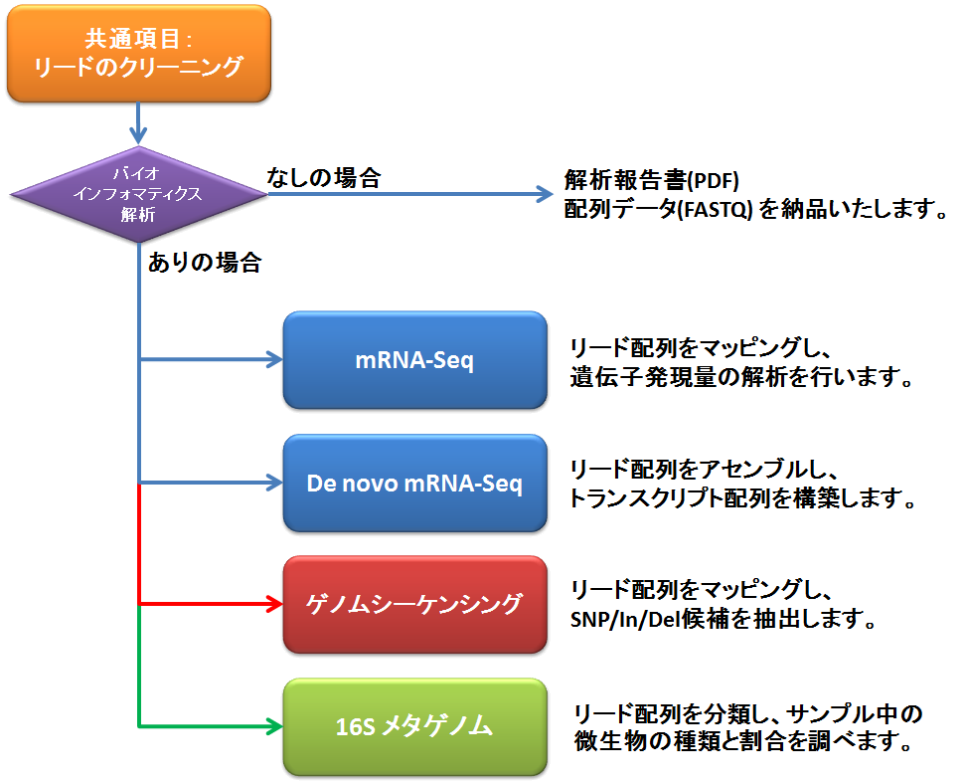
バイオインフォマティクス解析
シーケンシングで得られたリードのクリーニングを行い、アプリケーションに応じたバイオインフォマティクス解析を行います。
▼ 共通 ▼ mRNA-Seq ▼ De novo mRNA-Seq ▼ ゲノムシーケンシング ▼ 16S メタゲノム ▼ お見積もり・資料請求
共通
○クオリティーチェック(クリーニング前)
クリーニング前のリードのクオリティチェック
- リード補正度(QV値)、duplicate率、アダプターが無くなったか、ATGCの割合をチェックします。
- Blastなどを用いて、ご指定頂いた生物種由来の配列データが出力されていることを確認します。
(※ご希望の場合は、クオリティーチェックは行いません。)
○リードのクリーニング
- アダプター配列、クォリティーの低いリード及び塩基を除去します。
○クオリティーチェック(クリーニング後)
- クリーニング後のリードのクオリティチェック
- リード補正度(QV値)、duplicate率、アダプターが無くなったか、ATGCの割合をチェックします。
mRNA-Seq
<参照配列:mRNA配列の場合>
○マッピング
- BWAを使用し、クリーニング後のリード配列をmRNA配列にマッピングします。
○遺伝子発現量解析
- マッピング統計結果の算出
samtoolsを使用し、bamファイルからマッピングの統計結果を算出します。
発現量の補正はedgeRを用いて行います。
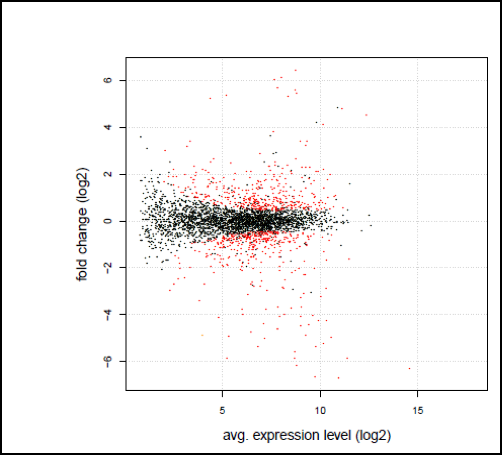
- 発現量比較解析
補正されたマッピングの統計結果を基に、サンプル間で各遺伝子発現量の変化があるかどうかを検定いたします。
<参照配列:ゲノム配列の場合>
○マッピング
- bowtieおよびtopphat(もしくはhisat2)を使用し、クリーニング後のリード配列を参照ゲノム配列にマッピングします。
○発現量の算出
- cufflinksもしくはstringtieを使用し、トランスクリプトのアッセンブルおよび発現量の算出を行います。
○遺伝子発現量解析
- トランスクリプトの結合
cuffmergeを使用し、cufflinksより得られた全サンプルのトランスクリプトを結合します。
- 発現量比較
cuffdiffを使用し、1により得られたトランスクリプト情報をもとに、各サンプル間の発現量比較を行います。
De novo mRNA-Seq
○De novoアセンブル
- velvetとoasesを使用し、De novoアセンブルを行い、コンティグ配列とトランスクリプト配列を生成します。
○トランスクリプト配列のクラスタリング
- cd-hit-estを使用して、トランスクリプト配列のクラスタリングを行い、冗長な配列を除きます。
ゲノムシーケンシング
○マッピング
- マッピング
BWAを使用し、クリーニングにより得られたリードを参照ゲノム配列にマッピングします。
- 重複リードの除去
Picard toolsを使用し、マッピングした結果からPCR増幅等のアーチファクトにより生成したと推定されるリードを除去します。
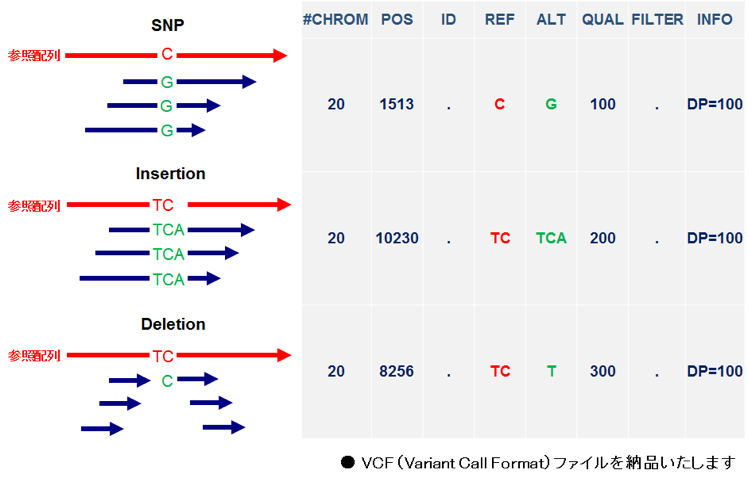
○【標準解析】SNP/In/Del候補抽出
- バリアントコール
samtoolsを使用し、マッピング結果から参照配列と異なる塩基を抽出します。
- フィルタリング
samtoolsを使用し、コールされたバリアントの内、設定値を満たすバリアントを抽出します。
- VCF(Variant Call Format)の作成
染色体名、位置、ホモ/ヘテロ別などの簡易なアノテーション情報を加えた変異部位候補の情報ファイルを作成します。
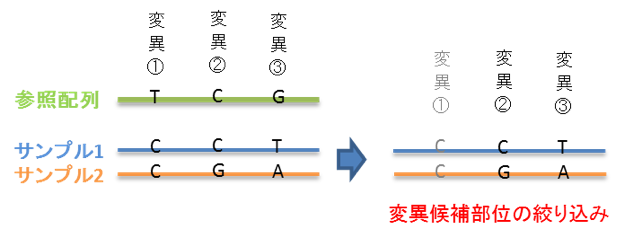
○変異部位の絞り込み解析
「【標準解析】SNP/In/Del情報抽出」では、各解析サンプルと参照配列間の 「1対1」 の比較による変異部位候補のリストアップを行います。
「変異部位の絞り込み解析」では、解析を行ったサンプル間の変異部位候補の情報のみを抜き出し、参照配列とサンプル配列の間の配列の相違を除きます。
○変異部位のアノテーション付与サービス
「【標準解析】SNP/In/Del情報抽出」では、ミニマムなアノテーション情報(染色体名, 位置, ホモ/ヘテロ別など)のみが付与されます。
「変異部位のアノテーション付与サービス」 では、変異候補部位の詳細なアノテーション情報を付与します。
詳細な情報には、遺伝子名や SNP ID、アミノ酸置換の有無といった情報が含まれます。
アミノ酸置換を伴う変異のみ抽出したい場合などに効果的なサービスです。
※データベース上に変異情報が豊富に整備されている生物種のみ、「変異部位のアノテーション付与サービス」が実施可能となります。
<デフォルト>
- 染色体名
- 位置
- ホモ/ヘテロ....
<アノテーション付加サービス>
- 遺伝子名(Gene ID)
- SNP ID
- アミノ酸置換の有無....
↑追加!
16S メタゲノム
○クラスタリング
リード配列のクラスタリングを行い、OTUを取得します。
※OTU (Operational Taxonomic Unit):類似性のあるリード配列をまとめたものです。
○種分類プロファイリング
QIIMEを用いたblastベースの解析を行い、OTUごとに細菌叢の分類、構成比などについてプロファイリングを行います。

お問い合わせ
サービス詳細やご不明な点などお気軽にお問い合わせください。
ユーロフィンジェノミクス株式会社
次世代シーケンス プロダクトサポート
E-mail ngs-jp@eurofins.com
TEL 03-6631-0106
▲ ページ先頭へ戻る